
这篇博客介绍MECAT这个软件,是中山大学的肖传乐老师2017年发表在Nature Method上的工作,用于三代Pacbio测序的基因组组装。MECAT的主要创新点在于前面提出了一系列针对Pacbio long read的序列比对方法,所以使得后续的组装非常迅速。Heng Li的minimap2是2018年发表的,是针对long read的比对开发的,也是目前应用非常广泛的一个long read比对软件,Accurate detection of complex structural variations using single-molecule sequencing(也就是比对软件NGMLR)这篇文章中,我搜索到了minimap2和MECAT的数据对比,首先看一下二者的运行情况
| Program | Wall clock (seconds) | RAM (byte) | Threads |
|---|---|---|---|
| minimap2 | 546 | 10051424000 | 10 |
| MECAT | 1236 | 22935784000 | 10 |
Table.1 Runtime comparisons over NA12878 Pacbio 1X 1.
下面的数据使用模拟数据来评估软件检测染色体结构变异(SV)的能力,这里模拟了不同长度,不同结构变异类型(插入、缺失、倒位、易位、重复等)的reads,然后将其比对到参考基因组,如果识别的SV,误差在10bp以内称为“precise”,如果误差大于10bp,称为“indicated”,因为read的模拟类型很多,这里我只选取了平均值来比较,具体的信息可以参见文章1.
| Program | AVG_precise | AVG_indicated |
|---|---|---|
| minimap2 | 52.7780707 | 16.65729014 |
| MECAT | 9.762036533 | 20.48438245 |
Table.2 Mapping simulated reads to human reference genome (GRCh37)1.
从这两个指标来看minimap2强于MECAT,二者在模拟数据上的比对错误率比较信息我没有在文献中搜索. 如果有人测试了,可以邮件告诉我结果,非常感谢!
Introduction
之前的方法,思路都是先寻找匹配的${k}$-mer,再进行local alignment进行延伸,但是local alignment往往计算时间很高,同时因为候选的位置比较多(尤其在基因组的重复位置),所以运行缓慢,因而基因组组装也往往非常缓慢.
文章开发了一种pseudolinear比对打分算法来过滤多余的比对,因此可以加速比对的过程.
Methods
Indexing and matching of reads
将目标read进行indexing,具体地,目标read的所有${k}$-mers都作为hash表的key,将read切分成一些Block,长度为${B}$,通常长度为${1000bp - 2000bp}$,hash表的value是${k}$-mer在read中的Block里的位置. 查询read也拆分成长度为${B}$的Block,并进行${k}$-mer的取样,用长度为${k}$的滑窗对每个Block进行取样,步长step length ${sl}$ 默认为${10}$,也就是说取样了大约${1/sl}$比例的${k}$-mer. searching block(查询read的Block)被视为matching,当且仅当他们的重叠的${k}$-mer大于阈值${m}$,两个read被视为matching,当且仅当至少有一个Block被matching. 这一步的过程见Fig.1
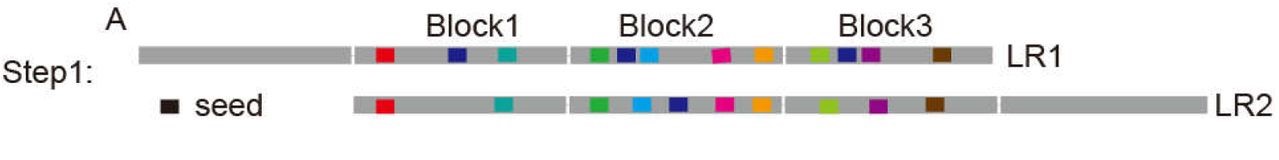
Fig.1 Alignment of k-mers between the blocks of two long read2.
Filttering by DDF
使用distance difference factor score(DDF)来过滤false matched read.
Mutual scoring
对于每一个matched read pair,首先随机选取一个matched block pair,并进行标记,然后给每一个matched ${k}$-mer打分,令${p_i,p_j}$表示该Block的第${i}$个和第${j}$个${k}$-mer,用 ${p^{\prime} _{i},p^{\prime} _{j}}$ 表示这两个${k}$-mer在相应的mathced pair Block的位置.
然后计算第${i}$个和第${j}$个${k}$-mer之间的${DDF_{i,j}}$
如果${DDF_{i,j}<\varepsilon}$,则表示这两个${k}$-mer相互支持,则这两个${k}$-mer的打分都增加${1}$,默认${\varepsilon=0.3}$. 在这里只使用非重复的${k}$-mer pairs进行打分,也就是说如果某个${k}$-mer在一个Block里面匹配了两个位置,则忽略这个${k}$-mer. 一个具体例子见Fig.2
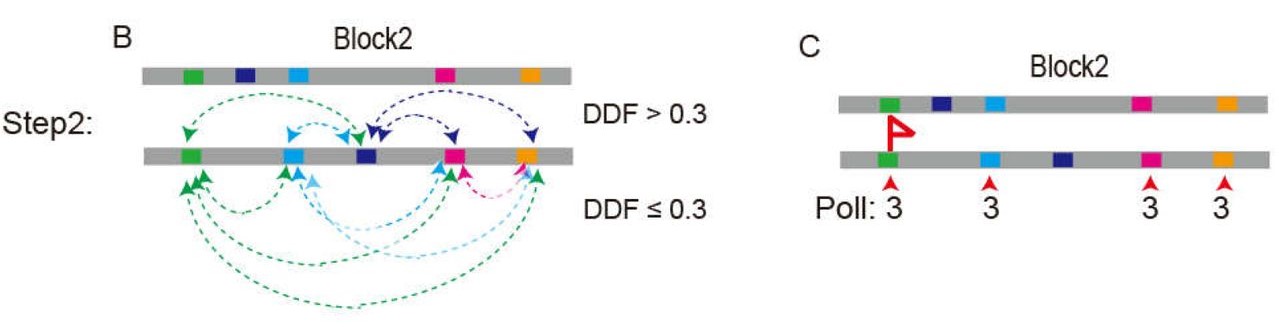
Fig.2 Scoring k-mer pairs in each block pair using DDF2.
如果有一个${k}$-mer是最高分,且超过阈值,那么我们选取该${k}$-mer作为进一步alignment的seed position,如果很多${k}$-mer打分一致,则随机选择一个作为seed. 例如Fig.2中选取了第一个标记为绿色的${k}$-mer作为seed.
Extension scoring
接下来从刚才选中的Block pair开始向它的邻居Block延伸打分,考虑neighbor block pair中的每个匹配的${k}$-mer,计算其与seed ${k}$-mer的DDF值,如果${DDF<\varepsilon}$,则seed ${k}$-mer的值加一,如果neighbor block中${80\%}$的overlaping ${k}$-mer满足${DDF<\varepsilon}$,则我们标记这个Block,并且不需要给这个Block中的${k}$-mers打分. 如果仍有未标记的Block,则进行Mutual scoring步骤,迭代进行这两步.
Pairwise alignment
选取Block长度为${2000bp}$,在对${k}$-mer进行打分之后,我们使用top-rank的${k}$-mer作为seed,来执行local alignment,如果overlap大于${2000bp}$,且overlap区域的错误了小于两倍的SMS read错误率(约${ 15\%}$),则视为match.
Alignment to reference
选取Block长度为${1000bp}$,取样步长${sl=20bp}$,如果还有一些read没有比对到ref上,则选取Block长度为${2000bp}$,取样步长${sl=10bp}$,也就是放松条件,并且增加取样再比对一遍(这样做的目的是因为第二步用时更长,所以筛取两遍),然后类似pairwise alignment,继续进行local alignment.
Correcting and Assembly
将其余read与目标read进行alignment,然后进行错误矫正. 然后至于基因组组装,有两种pipeline,可以直接将MECAT矫正的read输入Canu(称为MECAT-CA pipeline),或者用MECAT对correted read进行pairwise alignment之后,将比对结果输入Canu(称为MECAT pipeline).
Summary
总结来讲,MECAT主要是进行了快速准确的序列比对,这加速了错误矫正和read延伸的过程,而其序列比对的主要思想,是先使用Block的方法,过滤掉一批read,然后用${k}$-mer打分的方式,本身上是在寻找两个序列比对的靶心,然后用local alignment进行进一步的比对.