
这篇博客向大家介绍三代基因组组装软件Flye,这是Pavel A. Pevzner在2019年发表在nature biotech上的工作. Flye通过构建repeat gragh来表示基因组,同时在解决repeat问题上提出了一个非常好的思路.
Introduction
关于repeat graph,大家可以参考这篇博客文献分享:De Novo Repeat Classification and Fragment Assembly,关于三代测序的基因组组装问题,大致有两种思路,一个是“correction then assembly”,一个是“assembly then correction”,Flye采用的策略是先拼接后矫正,我想原因在于不矫正read,保留read的原始信息,才有可能解决unbridged repeat. 具体的原因,请在Methods部分细细品味.
Methods
为了更细致的理解Flye的思想,以及保持一些术语的统一,再这里我仍然按照Flye的行文思路进行讲解,当然一些思路和结论我们在博客文献分享:De Novo Repeat Classification and Fragment Assembly已经提及. Flye在方法部分,仍然是先介绍repeat characterization problem,然后引出repeat graph来解决repeat characterization problem,最后再给出repeat graph和assembly之间的关系.
Repeat characterization problem
考虑有向图${G}$和图中的一个长度为${n}$的tour ${T=v_1,v_2\cdots,v_n}$,我们称${i}$th和${j}$th顶点是等同的,如果他们对应图中的相同顶点,即${v_i = v_j}$. 这些等同的点${(i,j)}$,我们可以将其绘制在一个二维的网格中,我们称这个网格图为tour ${T}$的repeat plot ${Plot_T (G)}$. 如果已知${G,T}$,得到repeat plot ${Plot_T (G)}$是一个非常容易的问题.
而我们更关注这个问题的反问题,任给一个${Plot}$,来寻找图${G=G(Plot)}$和tour ${T}$.
基因组的dot-plot其实是一个矩阵,可以图表化genome中的所有repeat. 在repeat characterization问题中,我们关注于genome自身的local-alignment,那么每个一self-alignment对应基因组两个连续的repeat片段(segment)${x,y}$,${x}$和${y}$称为该alignment的${spans}$. 给定一个长度为${n}$的genome,以及自身比对的${Plot}$,Repeat characterization problem等价于构建图${G}$以及长度为${n}$的tour ${T}$(基因组的每个segment对应tour ${T}$中的subpath),其中${Plot=Plot(G)}$,tour ${T}$是符合alignment结果的.
Generating repeat plot of genome
Flye生成基因组的所有self-alignments,然后组合成repeat plot ${Plot}$.
Constructing a punctilious repeat graph
令 Alignment = Alignment(Genome,minOverlap),是基因组’Genome’中充分长的self-alignment的集合(长度最少是’minOverlap’). Flye设置’minOverlap’为read-set的N90(在这篇文章的数据集中,N90大致是${3000-5000bp}$)
给定了’Genome’的self-alignment的集合’Alignment’之后,接下来我们构建punctilious repeat graph,即一丝不苟的repeat graph RepeatGraph(Genome,Alignment). 得到的方法就是将’Alignment’比对上的positions ‘gluing’到一起. 然后将分支顶点之间的simple path收缩为一条边,这条边的长度等于原始path的边的数量. 这个图就是punctilious repeat graph,我们按照阈值${d=500}$,将边分为long和short.
之所以这样做,是为了下面简化punctilious repeat graph做铺垫,punctilious repeat graph在真实的基因组中会非常复杂,’gluing’操作会产生许多branching point.
From punctilious repeat graph to repeat graph
在比对的时候,repeat的端点很难完全对齐,那么在’gluing’之后,就会产生复杂的结构,比如分支顶点和short edge(${<d=500}$). repeat graph RepeatGraph(Genome,Alignment,${ d }$)定义为将punctilious repeat graph的short read收缩之后的图. 具体的收缩操作,就是将边的endpoint进行’gluing’,然后移除loop-edge. 考虑基因组tour,穿过repeat graph多次的就是repeat edges,一次的称为unique edge.
Approximate repeat graphs
在FragmentGluer中,花了很多的经历来处理Whirl/Bullbe,但是在三代测序中这些处理的很多参数很难抉择,因此我们采用下面的方法进行处理,只关注于self-alignment的端点.
考虑punctilious repeat graph中导致repeat graph中的branching vertices的顶点集合${V}$,令Breakpoints=Breakpoints(Genome,Alignment,${d}$),是导致${V}$的基因组的位置集合. 在Fig.1中,${V={A,B,D,E}}$,Breakpoints=${{1, 2, 4, 5, 7, 8, 9, 10,12, 13}}$.
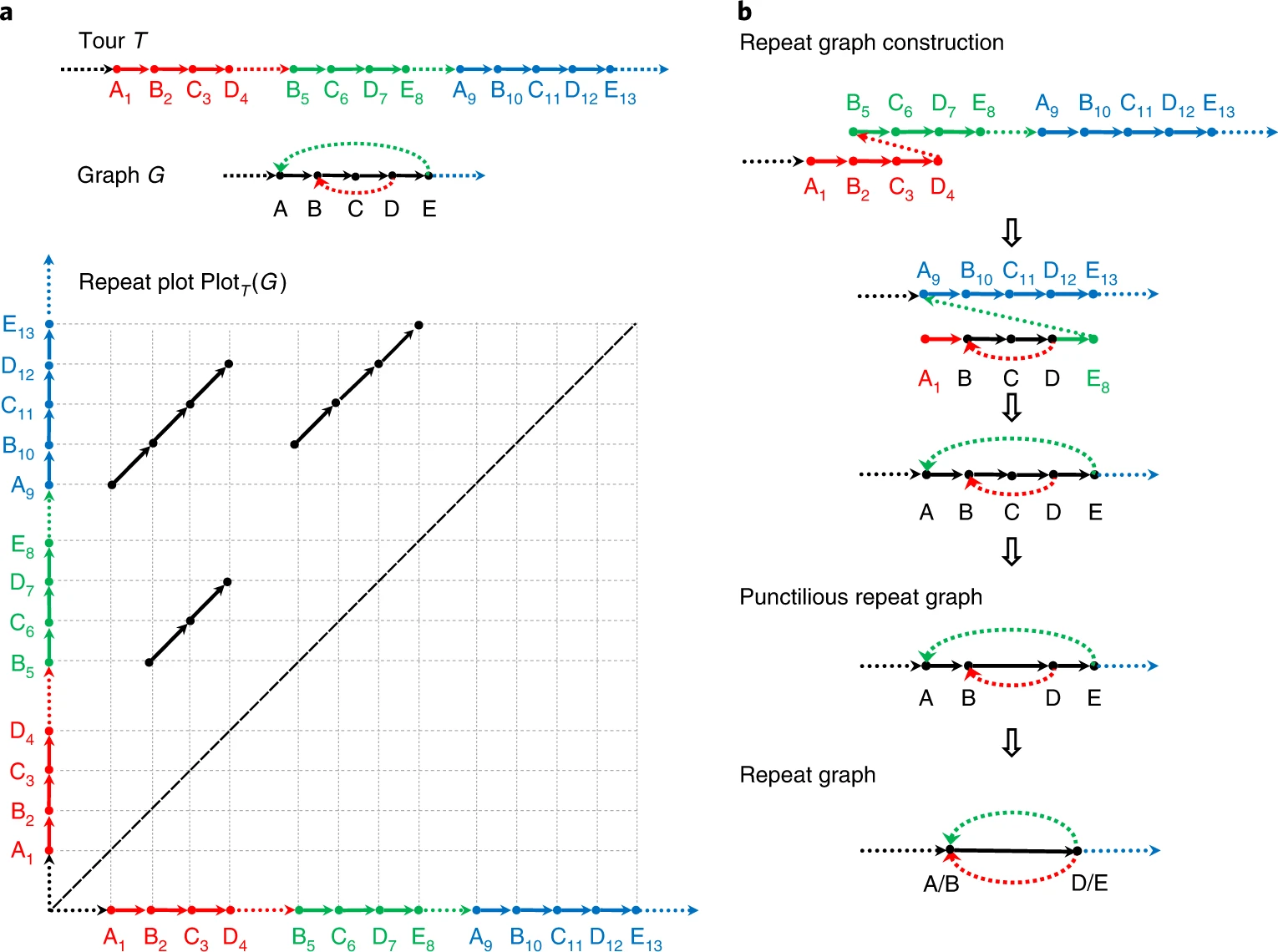
Fig.1 Workflow of constructing repeat graph1.
Flye选取’Alignment’中的endpoints在主对角线上的投影点来近似Breakpoints集合. 两个endpoint被称为close,如果他们在主对角线上的两个投影有一对是小于阈值${d}$的(允许一个水平投影,一个竖直投影).然后,Flye将endpoints按照close的关系,进行聚类. 聚成一类的endpoints被’gluing’到一起,进一步将平行边进行合并. Fig.2中(${d=0}$)作为示例,展示了Approximate repeat graphs的构建过程,同一个颜色的点就是被聚到一类的endpoints.
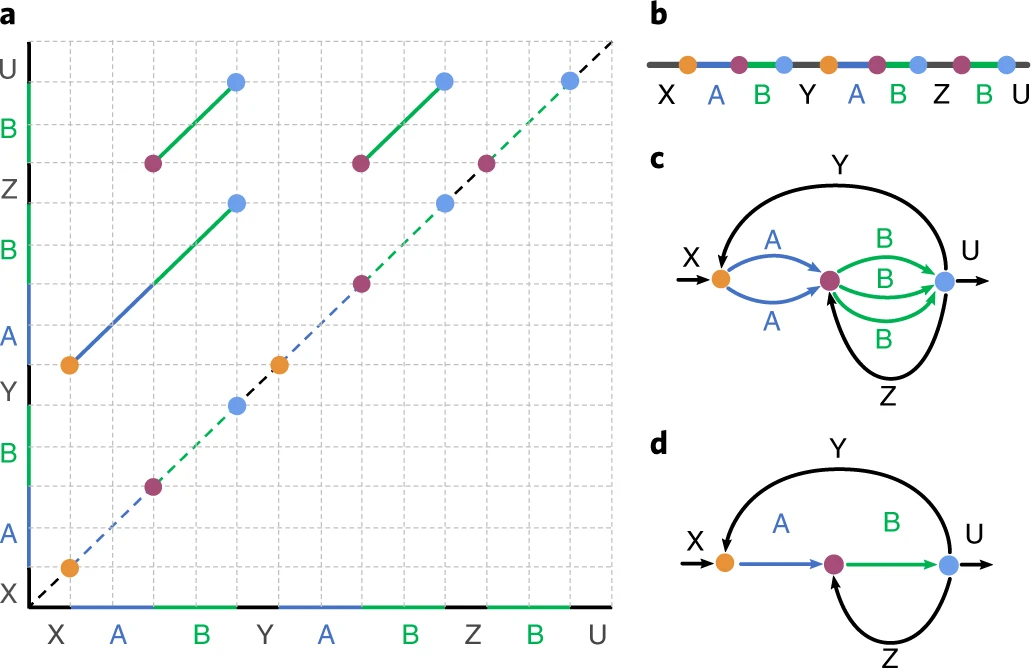
Fig.2 Workflow of constructing Approximate repeat graphs1.
Extending the set of breakpoints
之前我们提及过inconsistent alignment,理论上,${m}$个重复片段应该形成${\tbinom{m}{2}}$个比对,但是由于各种原因,${m}$个重复片段有一些两两比对没有被捕获,这就产生了inconsistent alignment. 那么这将导致一些’Breakpoint’的缺失. 比如Fig.3所示,由于缺失了${B_2,B_3}$之间的比对,所以紫色的方块位置的点没有归入’Breakpoints’集合. 因此就没有鉴定出来其中${A_2+B_2}$的镶嵌结构.
所以要进行’Breakpoints’集合的扩充,一个alignment-path中的点称为合法的,如果其两个投影点(水平、竖直)都属于’Breakpoints’集合,如果仅有一个,则称为非法的. 如果’Breakpoints’集合全部是合法点就称为合法的,否则称为非法的. Flye通过迭代的方法,试图加入最少的positions,使得’Breakpoints’集合成为合法的. 具体操作是为非法点加入missing的投影点,直到’Breakpoints’没有非法点.如Fig.3所示,因为五角星点只有一个投影点属于’Breakpoints’,为非法点所以加入方块点后,’Breakpoints’集合成为合法的.
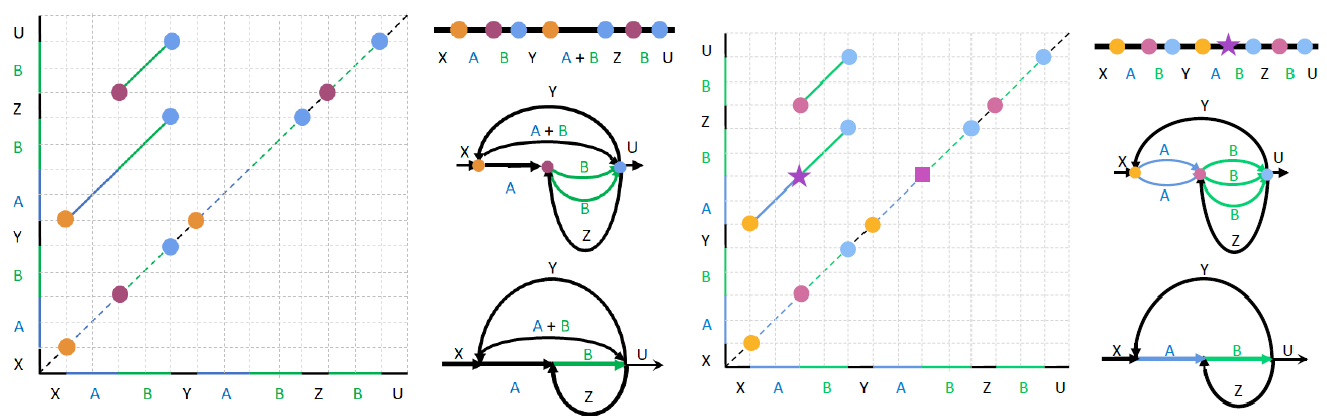
Fig.3 Extending the set of breakpoints1.
然后,和上面描述的一致,对’Breakpoints’集合中close的顶点进行合并,那么聚类的时候,其实在主对角线上,形成了segments(也就是cluster里面,位置最小和最大的之间的区域),因此这个新的集合称为’BreakpointSegments’,里面的元素称为segment. 两个segment是等价的,如果存在一个alignment-path的一个点,其两个投影分别落入两个segment中,如图Fig.4所示. 所以,通过这样,我们将segment进行聚类,那么合并Segment的时候,Flye选择只’gluing’ segment的middle points.
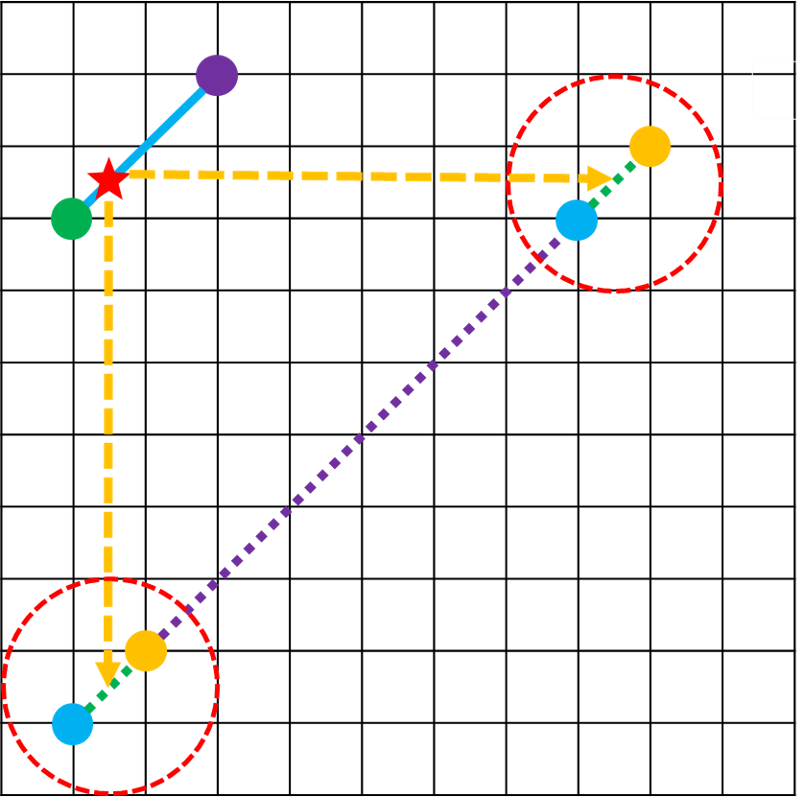
Fig.4 Equivalent segment.
Phased summary
我们简单梳理一下前面的思路,我们理论上需要从Genome,得到Alignment,然后构出punctilious repeat graph,再进一步得到repeat graph. 但是这样得到的repeat graph还需要进行复杂的去除Whirls/Bubbles的过程,因此我们选择只合并close的’Breakpoint’得到Approximate repeat graph,实际的操作上,我们使用’endpoints’的投影和’Extending breakpoints’操作得到’BreakpointSegments’,通过合并segment的middle points,再进一步如果平行边在’Alignment’中被比对,则进行合并来实现对于repeat graph的近似. 在后面的讨论中,我们简称Approximate repeat graph为repeat graph(RG).
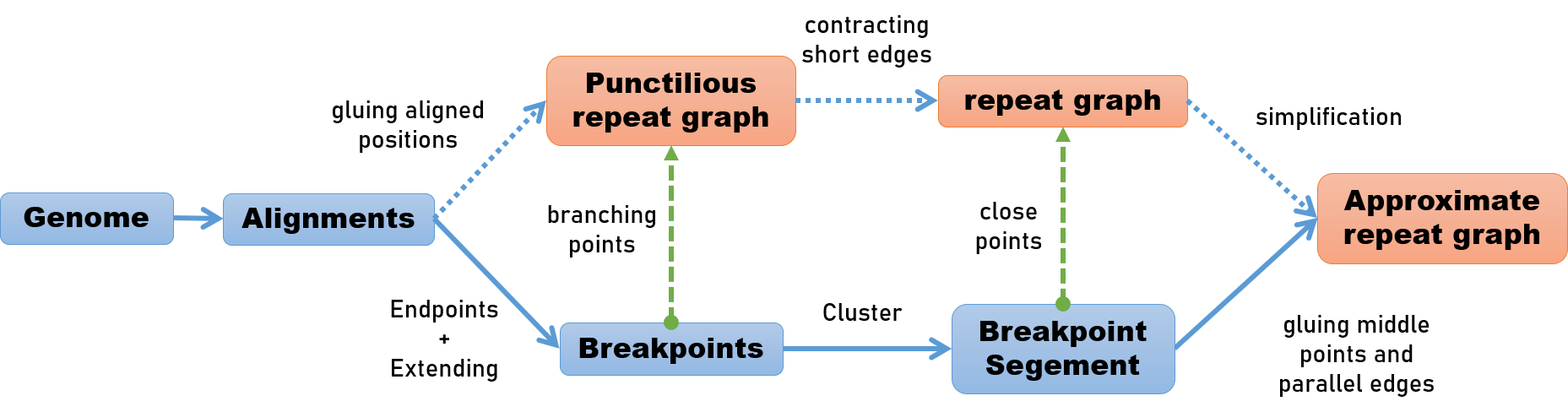
Fig.5 Phased summary.
Genome assembly
常见的拼接思路是,首先基于read构建assembly graph,再利用一些操作或者信息(例如piared-read)来解决repeat问题,最后输出contig. Flye认为更好的方法是继续基于contig构建assembly graph(这样的图会更简洁),然后进一步在这个图中去解决repeat问题. 这种思路的另一个好处是,容易结合一些二代三代数据混合拼接的思路.
ABruijn assembler构建contigs的方法,就是不断的延伸read,直到出现不能决定的地方(branching vetex in (unkonwn) repeat graph). read延伸的时候,需要至少‘minoverlap’的重叠.
Constructing disjointigs
令${UnprocessedReads=AllReads}$,从中任选一个read,然后进行延伸(保证至少‘minoverlap’的重叠),直到无法延伸,生成${ChainOfReads}$. 然后将${AllReads}$中的reads,全长比对到这个序列上,这一部分reads将被${UnprocessedReads}$集合中删除(这样可以加速disjointgs的构建过程). 然后,再用minimap2将read比对到${ChainOfReads}$上进行纠错,得到${ DisjiontigSequence }$. 这个过程,附录中说,可以将disjointig序列的错误率从${13\%}$提高到${1-5\%}$.
在延伸reads时候,其实Flye采用了并行的策略,所有${UnprocessedReads}$集合中的read都同时进行延伸,如果一个线程完成了,就和之前的disjointigs比对,如果和一个之前的disjointig重叠超过${10\%}$,就将这个disjointig删去,同时read释放回${UnprocessedReads}$集合.
Constructing assembly graph from disjointigs
然后将disjointigs按照任意顺序连接(用分割符隔开),得到序列${Concatenate}$,然后进行进一步得到’${Alignment}$’,按照Repeat characterization problem的描述,最终得到RepeatGraph(Concatenate,Alignments,${d}$). 这个RepeatGraph和基因组得到的repeat Graph是一致的,在博客文献分享:De Novo Repeat Classification and Fragment Assembly已经说明.
Resolving bridged repeats
Flye将reads比对回assembly graph,这里需要注意,对于每个repeat edge,Flye储存了多个拷贝(序列来自于原本的disjointigs). 所以Flye将read和所有这些拷贝都进行比对,选择一个最好的比对进行保留.
进一步,Flye首先计算整个grpah的平均测序深度${cov}$,然后将edge分为${low-coverage}$(深度小于${2\times cov}$),和${high-coverage}$(深度至少为${2\times cov}$). 然后再重新精确分类,刚才read比对回图上之后,我们可以得到read-path(read支持的path),一个边${e’}$被称为${e}$的successor,如果存在一个read-path${e’}$在${e}$的后面,对于${low-coverage}$且仅有一个succesor的边,被分类为unique,其余的边被分类为repetitive.
因为下面要介绍Flye怎么解repeat,所以这里先介绍一下bridged repeats和unbridged repeats(见图Fig.6),所谓bridged repeats就是指,存在一个read,能够跨过repeat区域,这样的话,我们在面对repeat区域的时候,就可以判定如何通过;如果不存在,就是unbridged repeats,那么处理起来就非常麻烦,有可能无法解开这个repeat.
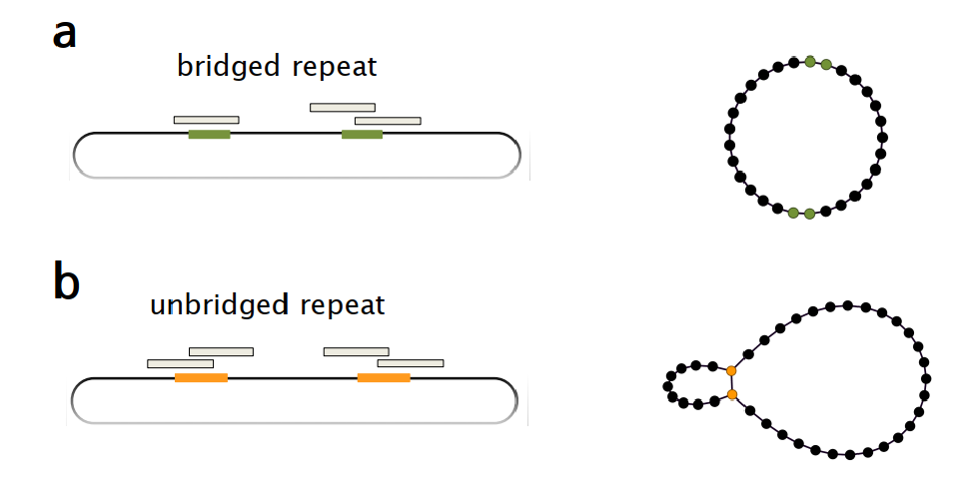
Fig.6 Bridged repeats and unbriged repeats2.
Untangling incident edges ${ e = (w, v) }$ and ${ f = (v, u) }$ in the condensed assembly graph amounts to substituting them by a single edge ${ (w, u) }$. 我觉得这一步的意思,是按照Fig.7进行操作.
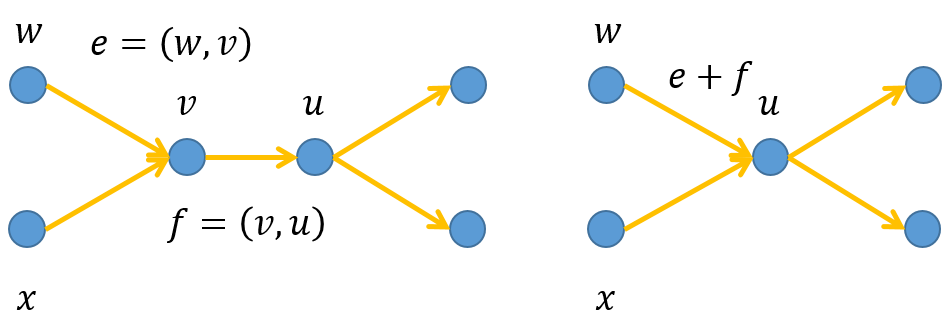
Fig.7 Condensed assembly graph.
因此,bridging read在condensed assembly graph被称为${(e,f)}$-read,如果其穿过两条连续的边${e,f}$的话. 我们在condensed assembly graph中定义一对偶联的边${e,f}$的‘transition${( e,f )}$’,其值为${(e,f)}$-read和${(f’.e’)}$-read之和(${e’,f’}$指的是${e,f}$的反向互补序列对应的边).
然后构建transiton graph,condensed assembly graph中的每个边${e}$,都对应两个顶点${e^h,e^t}$,也就是${e}$的head和tail顶点. 每个${(e,f)}$-read对应transition graph的一个无向边${{e^t,f^h}}$,权重等于transition(e,f). 在transition graph中,Flye调用最大权匹配算法,寻找到最大匹配之后,对于每个匹配${e^h,f^t}$,计算与${e^h}$或者${f^t}$邻接的边的总权重${TotalWeight}$,如果transition${(e^h,f^t)<TotalWeight/2}$,那么这个边将被忽略,因为支持这个路的read太少了. 最后Flye就这样迭代的解开一些边,再接着寻找最大权匹配,直到没有bridged能解的repeat.
Resolving unbridged repeats
Flye解决unbridged repeat的思路是利用不同的repeat区域之间,一般不会完全一致,会出现微小的变异(当然这里,其实还有个重要的条件,就是对于二倍体的生物,其repeat之间的差异大于两个同源染色体的差异,在文章的附录中,给出了具体的数据,repeat之间的差异在~${4\%}$,而同源染色体的SNVs大约是${0.1\%}$). Flye目前只能解决单倍体或者二倍体的二次重复的情况,换句话说,repeat的重复次数大于2,Flye是无法解决的.
具体地,Flye首先将read比对到REP edge的两侧,因此我们可以向前移动,分别构建一致性序列,这样就将repeat的区域缩短,迭代的进行,直到出现bridging read.
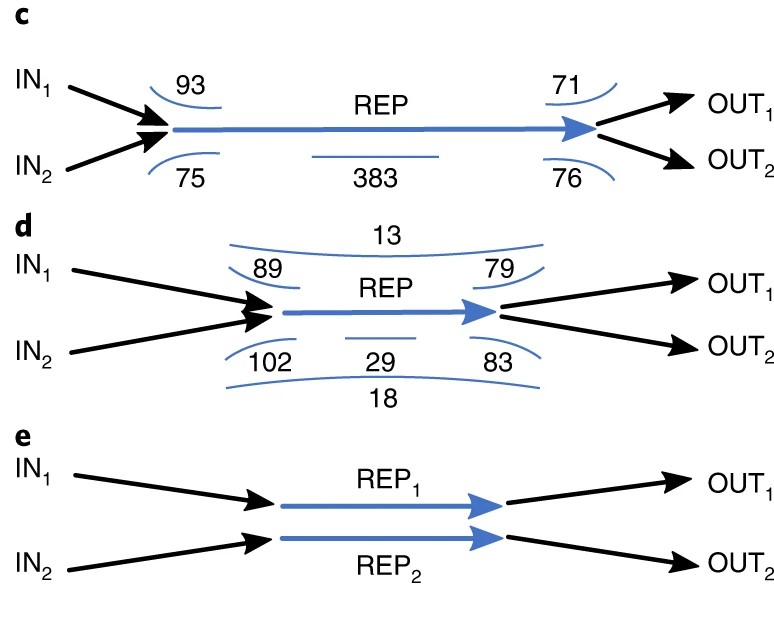
Fig.8 Resolving unbridged repeat1.