publications
2025
-
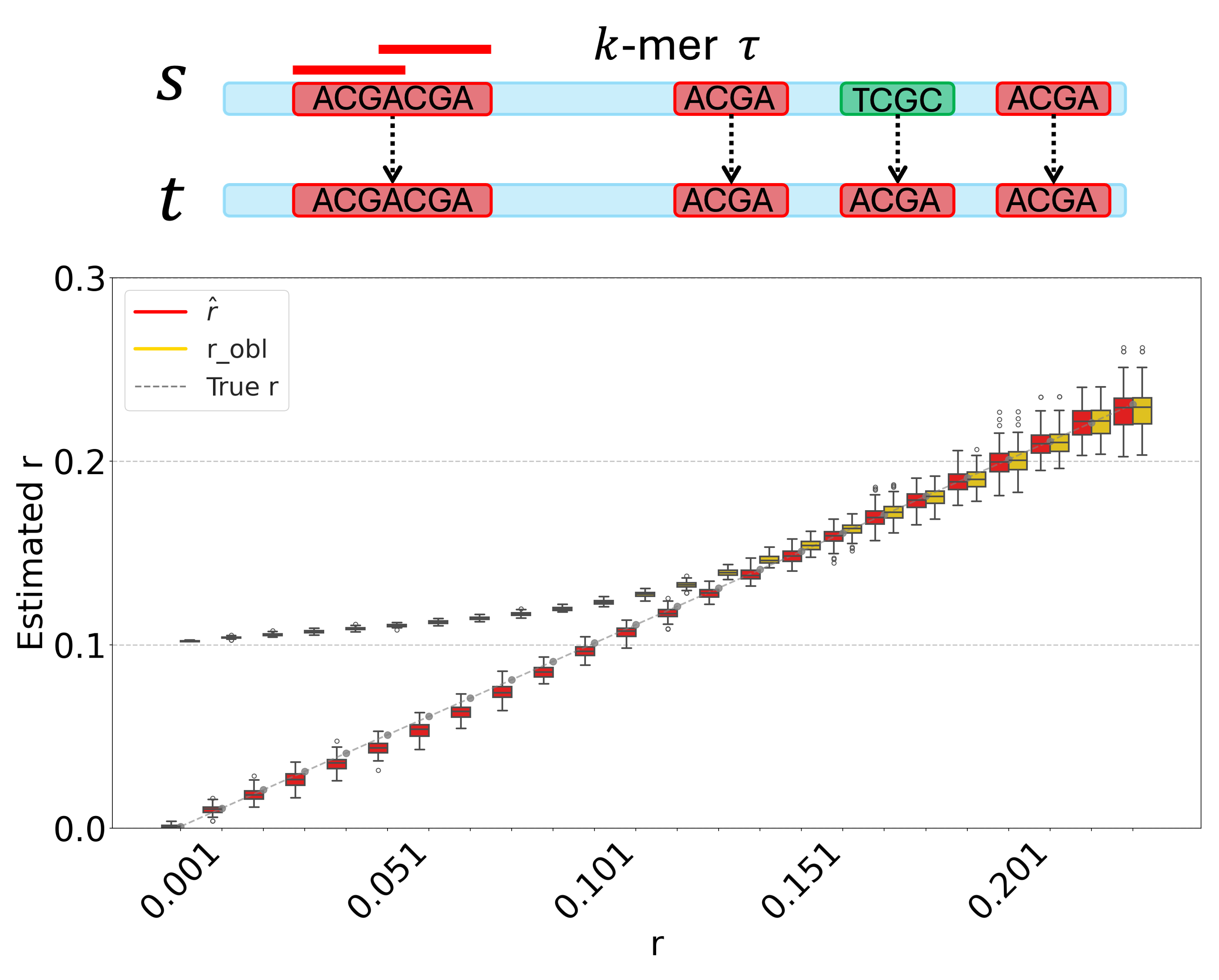 A k-mer-Based Estimator of the Substitution Rate Between Repetitive SequencesHaonan Wu, Antonio Blanca, and Paul MedvedevIn 25th International Conference on Algorithms for Bioinformatics (WABI 2025), 2025
A k-mer-Based Estimator of the Substitution Rate Between Repetitive SequencesHaonan Wu, Antonio Blanca, and Paul MedvedevIn 25th International Conference on Algorithms for Bioinformatics (WABI 2025), 2025K-mer-based analysis of genomic data is ubiquitous, but the presence of repetitive k-mers continues to pose problems for the accuracy of many methods. For example, the Mash tool (Ondov et al 2016) can accurately estimate the substitution rate between two low-repetitive sequences from their k-mer sketches; however, it is inaccurate on repetitive sequences such as the centromere of a human chromosome. Follow-up work by Blanca et al. (2021) has attempted to model how mutations affect k-mer sets based on strong assumptions that the sequence is non-repetitive and that mutations do not create spurious k-mer matches. However, the theoretical foundations for extending an estimator like Mash to work in the presence of repeat sequences have been lacking.In this work, we relax the non-repetitive assumption and propose a novel estimator for the mutation rate. We derive theoretical bounds on our estimator’s bias. Our experiments show that it remains accurate for repetitive genomic sequences, such as the alpha satellite higher order repeats in centromeres. We demonstrate our estimator’s robustness across diverse datasets and various ranges of the substitution rate and k-mer size. Finally, we show how sketching can be used to avoid dealing with large k-mer sets while retaining accuracy. Our software is available at https://github.com/medvedevgroup/Repeat-Aware_Substitution_Rate_Estimator.
- A k-mer-based estimator of the substitution rate between repetitive sequencesHaonan Wu, Antonio Blanca, and Paul MedvedevbioRxiv, 2025
K-mer-based analysis of genomic data is ubiquitous, but the presence of repetitive k-mers continues to pose problems for the accuracy of many methods. For example, the Mash tool (Ondov et al 2016) can accurately estimate the substitution rate between two low-repetitive sequences from their k-mer sketches; however, it is inaccurate on repetitive sequences such as the centromere of a human chromosome. Follow-up work by Blanca et al. (2021) has attempted to model how mutations affect k-mer sets based on strong assumptions that the sequence is non-repetitive and that mutations do not create spurious k-mer matches. However, the theoretical foundations for extending an estimator like Mash to work in the presence of repeat sequences have been lacking.In this work, we relax the non-repetitive assumption and propose a novel estimator for the mutation rate. We derive theoretical bounds on our estimator’s bias. Our experiments show that it remains accurate for repetitive genomic sequences, such as the alpha satellite higher order repeats in centromeres. We demonstrate our estimator’s robustness across diverse datasets and various ranges of the substitution rate and k-mer size. Finally, we show how sketching can be used to avoid dealing with large k-mer sets while retaining accuracy. Our software is available at https://github.com/medvedevgroup/Repeat-Aware_Substitution_Rate_Estimator.Competing Interest StatementThe authors have declared no competing interest.National Science Foundation, https://ror.org/021nxhr62, DBI2138585, OAC1931531National Institute Of General Medical Sciences, R01GM146462
2023
-
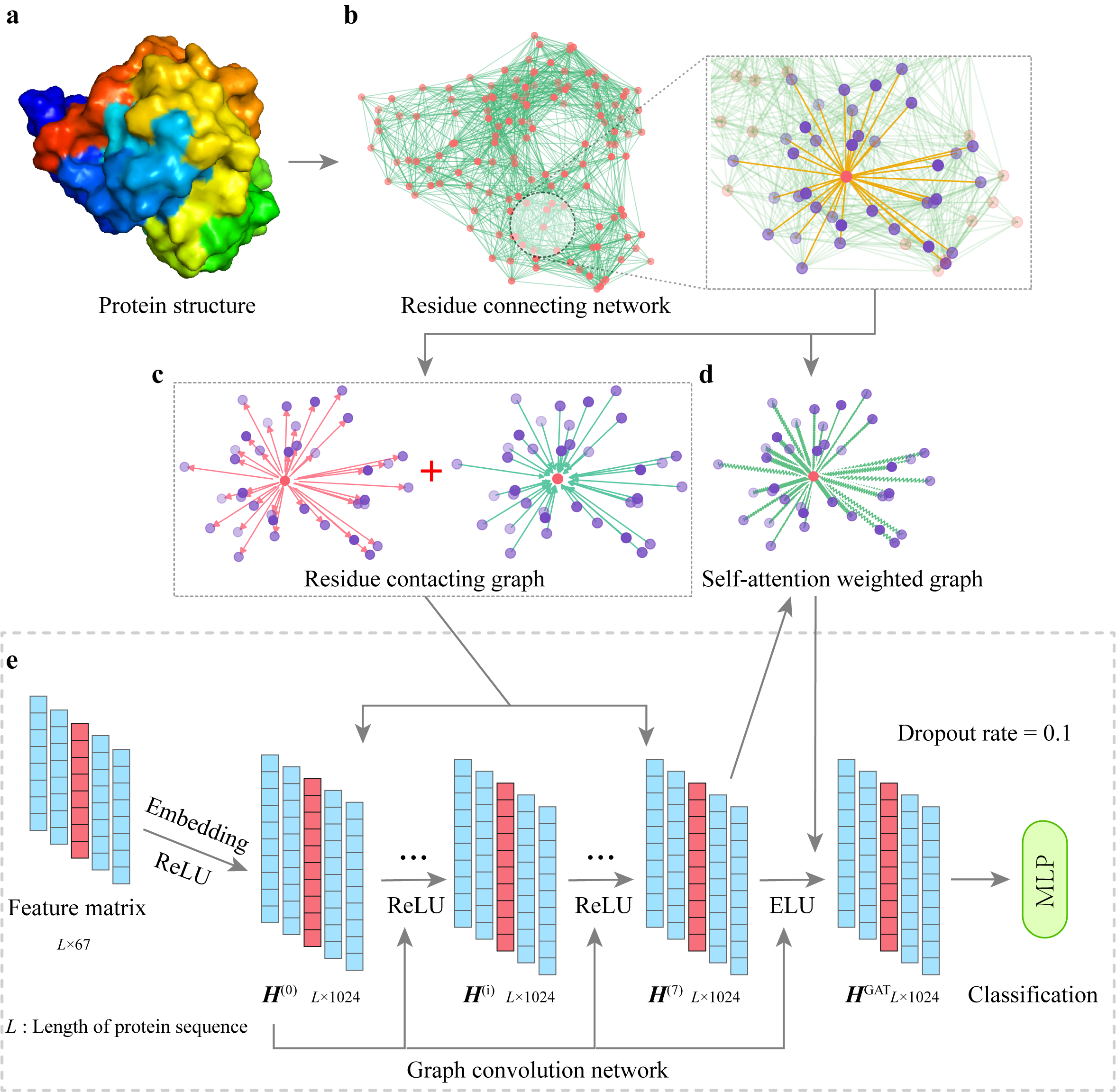 Spatom: a graph neural network for structure-based protein–protein interaction site predictionHaonan Wu, Jiyun Han, Shizhuo Zhang, and 3 more authorsBriefings in Bioinformatics, Oct 2023
Spatom: a graph neural network for structure-based protein–protein interaction site predictionHaonan Wu, Jiyun Han, Shizhuo Zhang, and 3 more authorsBriefings in Bioinformatics, Oct 2023Accurate identification of protein–protein interaction (PPI) sites remains a computational challenge. We propose Spatom, a novel framework for PPI site prediction. This framework first defines a weighted digraph for a protein structure to precisely characterize the spatial contacts of residues, then performs a weighted digraph convolution to aggregate both spatial local and global information and finally adds an improved graph attention layer to drive the predicted sites to form more continuous region(s). Spatom was tested on a diverse set of challenging protein–protein complexes and demonstrated the best performance among all the compared methods. Furthermore, when tested on multiple popular proteins in a case study, Spatom clearly identifies the interaction interfaces and captures the majority of hotspots. Spatom is expected to contribute to the understanding of protein interactions and drug designs targeting protein binding.