Haonan Wu (吴昊楠)
Department of Computer Science and Engineering, The Pennsylvania State University.

506, Wartik Lab
360 Science Drive
State College, PA, USA
Hi guys! I am a Ph.D. Candidate in Computer Science and Engineering at Penn State, under the supervision of Dr. Paul Medvedev.
Previously, I obtained my M.S. in Operations Research (OR) and B.S. in Biotechnology at Shandong University.
Research interests
I am interested in developing theories/algorithms/tools for bioinformatics and addressing biological problems via these methods. So, I aim to
- Design methods with solid math/algorithms or at least mathematical intuition.
- Build practically useful tools with heuristics benefiting the bio/bioinfo communities.
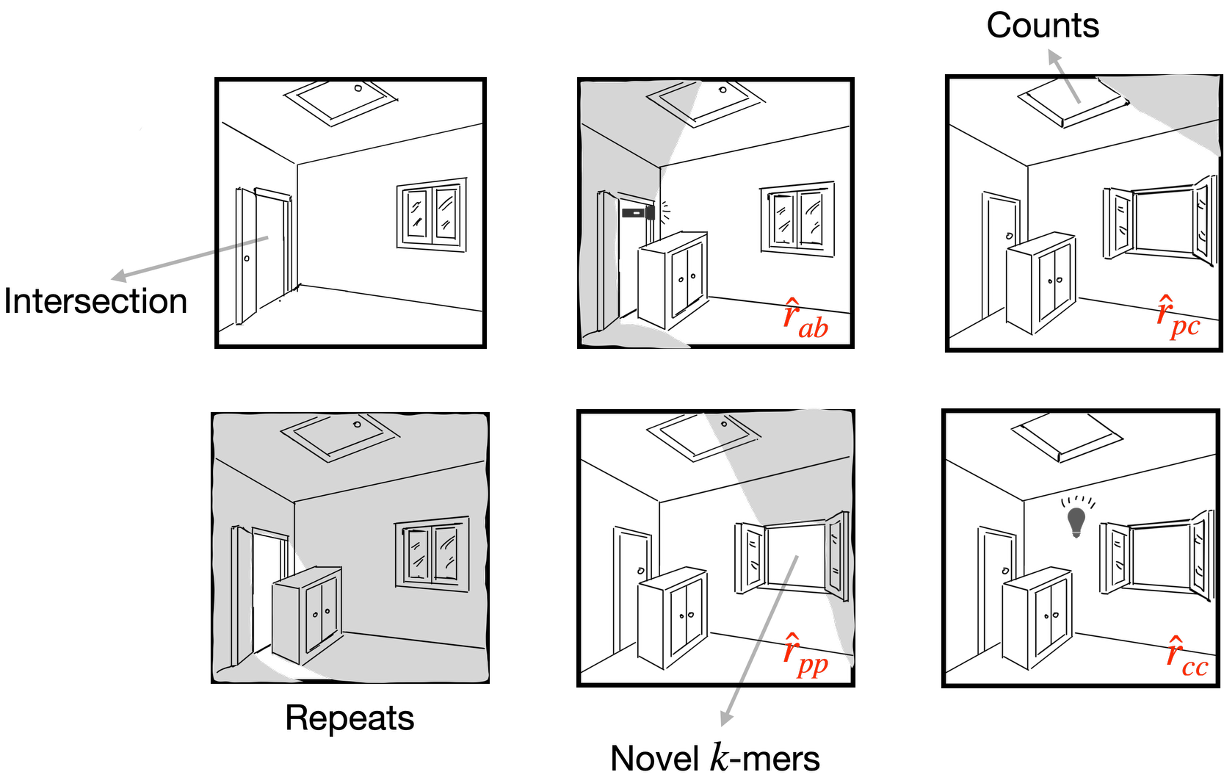
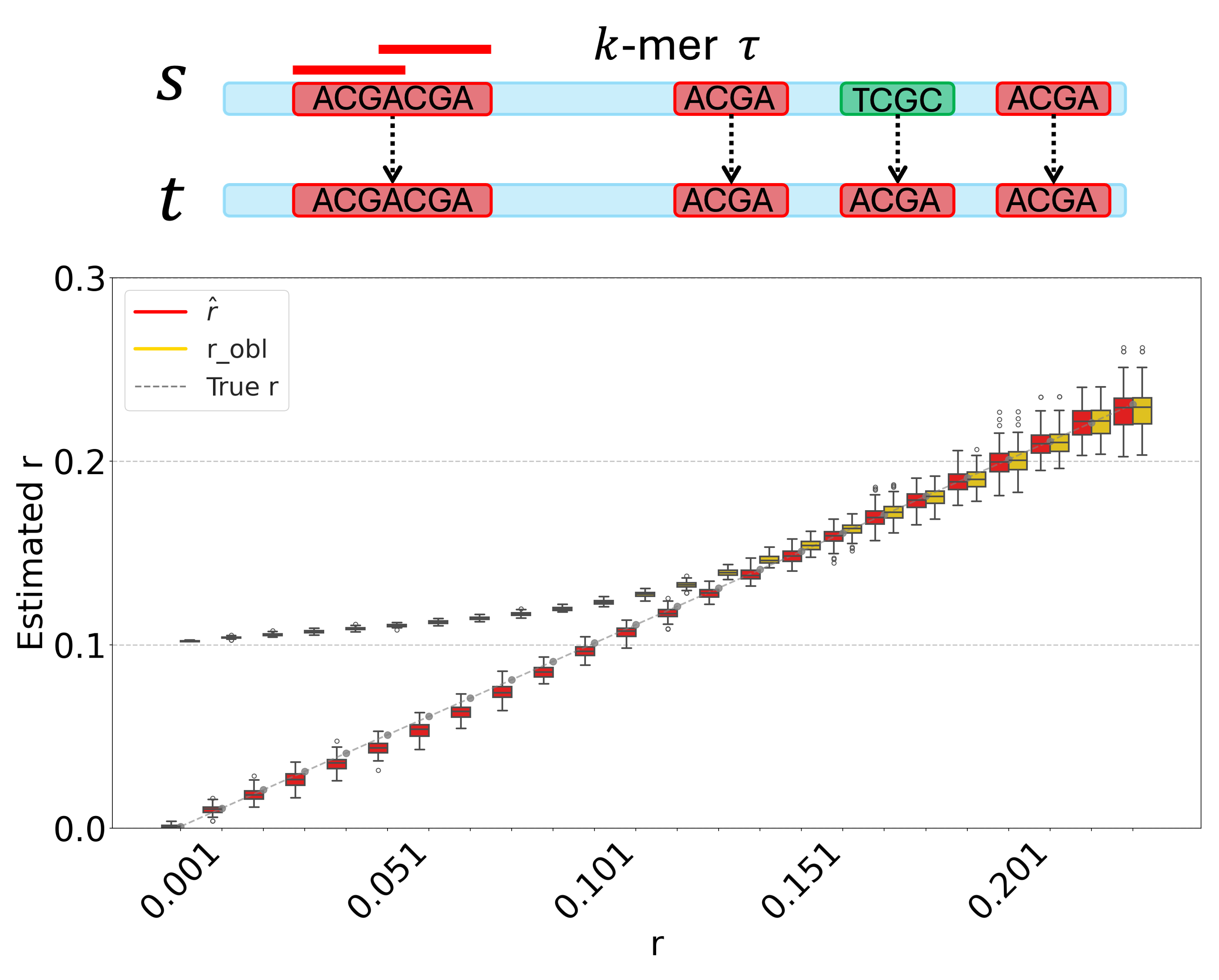
Currently, I am working on theories and estimators of mutation and evolutionary processes based on $k$-mers, particularly on repetitive genomic sequences.
news
| Apr 24, 2026 | I passed the Comprehensive Exam in the CSE PhD program at Penn State. 🍾 |
|---|---|
| Apr 02, 2026 | Our paper “The gift of novelty: repeat-robust k-mer-based estimators of mutation rates” at ISMB2026. 🎉 |
| Nov 06, 2025 | I give a talk at Genome Informatics 2025 with title “Don’t repeat no repeat: A k-mer-based estimator of the substitution rate between repetitive sequences”. |
| Oct 08, 2025 | I give the talk “Don’t repeat no repeat: A k-mer-based estimator of the substitution rate between repetitive sequences” at WABI2025 |
| Jun 14, 2025 | Our Paper A k-mer-based estimator of the substitution rate between repetitive sequences was accepted by WABI 2025. |